본 발명에 의해 복제 소의 생산이 가능하게 됨에 따라, 장차 축산학 및 의약학 분야에서
복제동물과 이를 이용한 약제 또는 장기 등 대량 생산하는 효과가 있다.
또한, 본 발명의 복제 소를 생산하는 기술은 소에 국한되지 않고 향후의 광범위한 변형된 시도에 따 라 이종간 동물들에도 적용될수 있는 효과가 있다.
그 뿐만 아니라, 본 발명에 따라 복제 동물을 생산하고 그와 관련된 응용분야가 개척될 수 있는 새로운 장이 열릴 수 있는 뛰어난 효과가 있으므로 본 발명은 축산학, 의약학 및 동물복제산업상 매우 유용한 발명인 것이다.
본 발명은 복제동물 및 그 생산방법에 관한 것으로서, 보다 상세하게는 핵이식(Nuclear Transplantation) 기술에 의해서 복제된 동물, 예컨대 복제된 소, 및 이 복제 소의 생산방법에 관한 것이다.
근래에 생명공학 또는 유전자 조작기술의 발달에 따라 유용한 작물들의 원하 는 형질들을 조합한 다양한 재조합 식물체들의 생산 성공사례가 발표되어 왔고, 최근에는 예컨대 양 등과 같은 복제 동물들의 생산에 성공한 사례들도 보고 되고 있다.
복제동물의 생산은 현실적으로 생명공학 분야에서도 실로 고도로 축적된 기술적 뒷바침이 전제된 후에야 비로소 가능한 것이므로, 이러한 점에서 상기 복제동물의 생산은 당해 기술 수준의 척도가 되는 것이다.
복제동물을 생산할 수 있는 능력은 장차 의약학, 축산학 등 산업분야에서 무한한 이용가능성이 잠재된 것으로 기대되고 있으므로, 복제동물의 생산은 기술혁신의 실로 획기적인 개가라고 할 수 있다.
이러한 복제된 소의 생산 방법의 기본 원리는 또한 다른 동물들을 복제하는데에도 응용될 수 있으므로, 향후 구체적 세부기술의 개발 여하에 따라 적용될 수 있는 동물의 범위도 넓어질 수 있을 것으로 전망된다.
따라서, 본 발명의 목적은 상기 핵이식 기술에 의해서 복제된 동물, 특히 복제된 소, 및 이 복제된 소를 생산하는 방법을 제공하는데 있다.
특허 기술 설명
본 발명자의 상기 목적은 난자의 체외성숙 및 핵이식 기술에 의해서 성숙난자의 탈핵 및 세포융합을거쳐 활성화시킨후, 후활성화 및 배양시킴으로써 본 발명의 목적이 되는 복제된 소를 생산함으로써 달성하였다.
본 발명에 따르면, 다음과 같은 공정단계들로 구성되는 복제 소의 생산방법이 제공된다:
(a) 도축난소로부터 미성숙난자를 채취 배양하는 단계;
(b) 소 신체의 다양한 조직으로부터 세포주를 분리하여 보존하는 단계;
(c) 상기 (a)단계에서 준비된 성숙난자의 투명대의 일부를 절개하고 세포질의 10∼30%를 제거하여 탈핵을 실시하는 단계;
(d) 상기 (b)단계에서 준비된 공여세포를 상기 (c)단계의 핵이 탈핵된 난자에 이식하는 단계;
(e) 상기 (d)단계에서 이식 및 밀착시킨 작성란을 DC 조건의 전압으로 전기융합시키는 단계;
(f) 상기 융합된 세포를 융합과 동시에 또는 화학물질이 첨가된 배지내에서 활성화시키는 단계;
및
(g) 상기 활성화된 세포를 후활성화 및 미소적 배지내에서 배양시키는 단계이다.
본 발명은 (a) 도축난소로부터 미성숙난자를 채취·배양하는 단계;
(b) 소 신체의 다양한 조직으로부터 세포주를 분리하여 보존하는 단계;
(c) 상기 (a)단계에서 준비된 성숙난자의 투명대의 일부를 절개하고 제 1극체와 함께 세포질의 10 내지 30%를 제거하여 탈핵을 실시하는 단계;
(d) 상기 (b)단계에서 준비된 공여세포를 상기 (c)단계의 탈핵된 난자에 이식하는 단계;
(e) 상기 (d)단계에서 이식 및 밀착시킨 작성란을 DC 조건의 전압으로 전기융합시키는 단계;
(f) 상기 융합된 세포를 융합과 동시에 또는 화학물질이 첨가된 배지내에서 활성화시키는 단계; 및
(g) 상기 활성화된 세포를 후활성화 및 미소적 배지내에서 배양한 후 검란하는 단계로 이루어진 복제 소의 생산방법에 있어서, 상기 (c)단계에서 투명대 일부를 절개하고 제1극체와 함께 세포질의 10~30%를 제거하여 탈핵하고, 상기 (e)단계에서 DC 조건의 전압이 0.75∼2.25kv/cm으로 1∼2회에 걸쳐 회수×시간이 총 10~60㎲으로 되도록 조정하여 전기융합하고, 상기 (f)단계에서 융합과 동시에 일렉트로퓨젼 방법으로 활성화를 유도하는 경우에는 Ca 2+ 첨가 융합배지 내에서 일렉트로퓨전을 수행하고, 이노마이신 처리법으로 활성화를 유도하는 경우에는 차광조건하에 50μL 점적내에서 4분간 정치한 35mm 디쉬내에 10% FBS 또는 30mg/mL BSA가 첨가된 세정용 매질하에서 이노마이신을 제거한 뒤 5분간 정치시킴으로써 세포융합된 세포를 활성화시키고, 상기 (g)단계에서 시클로헥시미드 25μL 점적 내에 5~10개의 작성란을 침지하여 6시간 배양하거나, DMAP 25μL 점적 내에 5~10개의 작성란을 침지하여 4시간 배양시킴으로써 후활성화 단계로 이루어진다.
본 발명의 바람직한 실시예에 따르면, 상기 (b) 단계의 세포는 소의 자궁관류액, 자궁내막, 난관에서 분리된 세포주이거나 난구세포일 수 있다.
본 발명의 다른 실시예에 따르면, 상기 (b) 단계의 세포는 소의 귀 또는 근육으로부터 분리된 세포이거나 태아섬유아 세포이다.
또한, 상기 (b) 단계의 보존처리는 계대배양, 혈청기아배양 또는 동결보존에 의해 이루어지게 된다.
본 발명의 다른 실시예에 따르면, 상기 방법중 어느 하나의 항의 방법에 따라 생산된 복제 소가 제공된다.
이하에서는 본 발명 방법에 따라 수행된 바람직한 실시예 및 실험예를 구체적으로 기재한다.
실시예 1 : 소의 체외수정(in vitro ferbilzation)
제1단계 : 체외성숙
체외성숙(In vitro maturation)을 위한 배지(Medium)는 다음과 같이 준비하였다.
배양(culture)용 TCM199(No. 3); 세척(washing)용-TCM199(No. 2);
혈청(FBS, 1ml씩 분주되어 있는 것); E 2 (No. 7); FSH(No. 7)
상기 배양액을 준비하여 4-웰(well) 디쉬(dish)에 0.5㎕ E 2 을 분주하고, 알코올을 휘발시킨다.
12.5㎕ FSH(NO.7)을 분주하고 FBS 1㎖를 배양용 TCM199 9㎖에 첨가한 후 각각의 웰에 0.45mL씩 분주한다. 스퀴즈 보틀(Bottle)을 이용하여 웰 사이에 증류수를 분주한다.
35mm 디쉬에 남은 TCM 199 2.5ml를 분주하여 최종 세척액으로 사용한다.
난소채취는 생리식염수(35℃)와 가위, RP장갑, 장화등을 준비하여 도축장에서 난소를 채취한다.
검란준비를 위하여는 1cm 간격의 격자 눈금을 그은 100mm 디쉬와 세척용 TCM199을 2ml씩을 분주한 35mm 디쉬를 준비한다.
채취한 난소로부터 18G 니들(needle)이 장착된 10ml 주사기를 사용하여 2∼6mm의 난포를 흡입한다.
다음에, 검란 및 세정을 수행한다. 먼저 난구세포가 충분히 부착되어 있고 세포질이 균질한 난자를 선발하고, 선발한 난자는 3회의 세정을 거쳐 최종 배양용 TCM199에 골라 놓고 각 웰 당 20∼50개의 난자를 35㎕씩 뽑아 분주한다.
체외성숙을 수행함에 있어서는 5% CO 2 배양기내에서 24시간 배양한다.
제2단계 : 체외수정
체외수정(In vitro fertilization)은 다음과 같이 배지와 과정을 거쳐 수행하였다.
배지 준비물은 Cap-TALP(뚜껑 열고 배양기, No. 1); IVF-TALP(뚜껑 닫고 배양기, No. 1); Washing-TALP(항온수조, No. 1); 헤파린(배양기, No. 8)로 하였다.
수정과정(Procedure)은 첫째, 미소적 작성 : 60mm 디쉬위에 43㎕ IVF-TALP 미소적(drop)을 만들고 인큐베이터(incubator)에 넣어둔다.
모든 미소적은 미네랄오일로 커버, 1개 미소적 당 5∼7개의 난자를 첨가함을 고려하여 적절한 수의 디쉬를 준비한다.
둘째, 스윔-업(Swim-up) : 5ml 둥근바닥 시험관(round bottom tube)를 10개 준비하여 cap-TALP를 0.8∼1ml씩 분주하고, 헤파린(Heparin)과 함께 배양기에 넣어 둔다. 성숙 22시간에 정액 스트로우(straw)를 2∼3개 녹이고 파스퇴르 피펫을 사용하여 튜브 바닥에 약 0.1ml씩 분주, 배양기에 1시간동안 넣어둔다.
셋째, 난자의 세정 : 스윔-업(Swim-up)을 하는 동안 3개의 35mm 디쉬에 세정용-TALP를 2ml씩 분주하여, 체외성숙중인 난자를 3회 세정한다. 세정한 난자는 작성한 각각의 미소적에 3㎕, 5∼7개씩 첨가한다.
넷째, 정자의 세정 : 스윔-업(Swim-up) 1시간후 정자 부유 상층액을 0.7∼0.8ml씩 흡입하여 15ml 튜브에 모은다.
모은 정자 부유액은 500∼700g, 6∼10분간 원심분리하고 100∼200㎕의 정자 펠릿(pellet)을 남긴 후 상층액을 제거하고 Cap-TALP로 1∼2회 더 원심세정한다.
최종원심분리 후 상층액을 제거하고 약 100㎕의 정자 펠릿을 남긴다.
다섯째, 정자농도의 결정: 4% HCl 190㎕와 정자 펠릿 10㎕를 섞은 후 혈구계산반의 양쪽에 적당량을 주입하여 100∼400배의 배율하에서 정자의 수를 센다. 5개의 큰 구획중 가운데의 구획에서(이 구획은 25개의 소구획으로 나뉘어져 있다) 5군데(네 모서리와 한가운데)에 있는 정자를 세고 나머지 한쪽 계산반의 것도 동일한 방법으로 센다.
이렇게 모두 10군데를 센 후, 총 100개가 넘으면 정자 펠릿의 부피(마이크로 피펫으로 100㎕전후로 조정하면서 측정한다.)
× 총 개수 ÷ 100을 한 수가 첨가할 헤파린의 양이고, 이 수치에서 펠릿의 부피을 뺀 수가 추가로 첨가할 cap-TALP의 양이 된다(예: 카운팅한 개수가 130개이고 펠릿이 90㎕이면 90×130/100=117이 첨가할 헤파린의 양이고 117∼90이 추가로 첨가할 Cap-TALP의 양이다.)
수정과정에서 수정능 획득을 위해 헤파린(최종농도 100㎍/ml)을 첨가하였으며, 15분간 배양기에서 배양하여 수정능 획득을 유도하였다.
또한, 정자를 4㎕씩 미소적내에 주입(최종 정자농도는 2×106개/ml)하고 5% CO 2 배양기내에서 18∼30시간 배양하였다.
제3단계 : 체외배양
체외 배양(In vitro cultivation)은 다음과 같이 준비하여 수행하였다.
배지(Medium) 준비물은 mTALP(incubator, No. 11); 세정용 배양액 (washing-TCM 혹은 washing-TALP)(waterbath)으로 하였다.
그 과정(Procedure)은 먼저, 미소적을 작성한다. 35mm 혹은 60mm 디쉬, 25∼30㎕ mTALP 미소적, 인큐베이터(incubator)에 보관(1개 점적 당 5∼10개의 난자를 첨가함을 고려하여 적절한 수의 디쉬 준비)하고, 35mm 디쉬 3개에 세정용 배양액을 2ml씩 분주, 가온판(warm plate)위에 올려 놓는다.
다음에, 난구세포제거(Denuding)과정으로 세정용 배양액이 들어있는 15ml 시험관(tube)에 체외수정된 난자를 넣어2분간 와동(vortexing)하거나 mouth pipetting하여 난구세포를 제거한다.
난구세포가 제거된 난자를 3회 세정, 5㎕에 5∼10개씩 흡입하여 미소적에 주입한 후, 5% CO 2 배양기 또는 5% CO2 , 5% O 2 , 90% N 2 에서 5∼8일간 배양하되, 체외수정 시작시기부터 계산하여 48시간 전후로 검란, 발육단계별로 구분하여 미소적내에 배치한다.
최종 검란은 수정 7일에 실시하여 후기배 발육율 계산한다.
제4단계 : 수정란 동결(Embryo Freezing, 기기-FHK, ET1N)
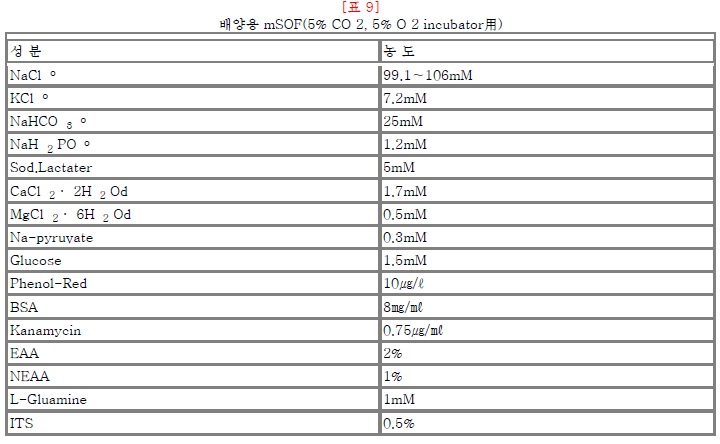
수정란 동결을 위한 동결배지(Freezing Medium)로 PBS(+)(표.9) 2.25ml와 혈청(FBS) 2.25ml를 35mm 디쉬에 함께 분주하여 희석한 후 글리세롤(Sigma, G-9012)을 0.5ml 첨가(10% 글리세롤)한다.
다음에는 동결기를 가동하여 -5℃를 유지시키고, 동결할 수정란을 선발하여 PBS(+)로 세정하고 동결배지에 넣어 15∼20분간 정치시킨다.
그후, 0.25ml 스트로우에 수정란을 장착(양 끝단에 공기층을 2층씩 만들고 가운데에 수정란이 포함된 배양액을 흡입, 봉인은 불에 달군 집게(forcep)를 사용하여 열밀봉(heat sealing)한다.
즉, 면봉-(배양액)-공기층-(배양액)-공기층-(배양액+수정란)-공기층-(배양액)-공기층-(배양액)-열 밀봉(heat sealing)으로 수행한다.
그리고, -5℃에서 동결기에 스트로우를 장착(면봉이 아래쪽)하고, 5분간 이 온도를 유지시킨 다음, 액체 질소탱크에 집게(forcep)를 10∼20초간 넣었다 빼어서 스트로우의 상부 2번째 공기층(밑줄그은 부분)의 하단을 살짝 집어주어 식빙(seeding)시킨다.
식빙 직후 0.3℃/min의 비율로 -30℃까지 온도를 하강시킨 다음 -30℃가 되면 10분간 온도를 유지시킨 후 액체 질소탱크내에 동결보존한다.
제5단계 : 동결란 융해(Thawing)
동결란을 융해하기 위해 다음과 같이 배지를 준비하였다. 6% 글리세롤, 3% 글리세롤 및 0% 글리세롤 용액; 수정란이식 배지(Embryo transfer medium)- PBS(+), FBS(1ml씩 분주된 것).다음, 동결란을 융해하기 위하여는, PBS(+) 4ml에 FBS를 1ml 첨가하여 20% FBS 용액을 제조한 다음, 융해 용액 및 20% FBS 첨가한 PBS(+)용액을 35mm 디쉬에 각각 분주하고
라벨링(labeling)한다.
그리고, 약 30℃의 온수를 준비(직경이 20cm이상인 용기사용)하여, 액체 질소탱크로부터 동결 스트로우를 꺼내 공기중에서 약 5초간 정치한 후, 온수에 약 30초간 담가서 융해(흔들거나 세우지 말 것)시킨다.
그 후, 6% 글리세롤 용액에 스트로우내의 수정란을 침적시키고 5분간 정치시킨 다음, 3% 글리세롤용액에 스트로우내의 수정란을 침적시키고 5분간 정치시킨 후, 0% 글리세롤용액에 스트로우내의 수정란을 침적시키고 5분간 정치시킨 다음, 수정란 이식배지(Embryo transfer medium)에 수정란을 침적시킨 후 스트로우에 장착하고, 수정란이식을 실시한다.
제6단계 : 난자 염색(Aceto-orcein 이용)
난자염색을 위한 준비물(Preparation)로서 고정액 (Carcoddylate buffer,조직절편고정용액)을 사용하고, DW, 에탄올, Aceto-orcein 용액(아세트산 45ml + orcein 1g + 증류수 55ml), 슬라이드 글라스(슬라이드 글라스), 커버 글라스(커버글라스), 바세린 파라핀 용액을 준비하였다.
난자염색과정(Procedure)은 다음과 같다.
즉, 슬라이드글라스의 1/3 부위에 4군데(커버글라스의 모서리에 닿을 정도) 파라핀 점적을 형성시킨 다음, 난자를 4군데의 점 가운데 마우스피펫으로 4∼10개 정도 올려놓고 커버글라스를 덮는다.
다음, 볼펜이나 피펫팁 등의 가는 물건을 이용하여 난자를 누른다.
그리고, 고정액을 흘려 보내고 거름종이를 반대편에 대어 고정액 통과 후 10∼15분간 정치(습윤유지)시킨 다음, DW를 통과시킨다.
그 후, 에탄올을 통과시킨 다음, Aceto-orcein 으로 통과하여 염색시킨다.
그리고, 현미경하에서 관찰한다.
제7단계 : 난자 및 수정란의 염색(Giemsa Stain 이용)
난자와 수정란의 염색을 위하여 다음과 같이 준비하였다.
저장처리용액 (0.9% 시트르산 나트륨 용액 또는 0.56% KCL 용액); 고정액Ⅰ(메탄올:빙초산:증류수 = 5 : 1 : 4); 고정액Ⅱ( 메탄올:빙초산 = 3 : 1); 5% Giemsa; 슬라이드 글라스. 염색과정(Procedure)은 다음과 같다.
수정란을 저장용액에 15분간 침지시켜 수화시킨 다음, 가능한 적은 용량의 저장액과 함께 수정란을 슬라이드 글라스 위에 점적시키고, 수정란위에 마이크로피 펫(micro 피펫)을 사용하여 고정액Ⅰ의 점적(zona pellucida를 제거)을 형성시킨다.
고정액Ⅱ를 점적하여 슬라이드 글라스에 고정시키고 공기중에서 건조시킨다.
Giemsa 용액내에서 15분간 염색하고,증류수를 이용하여 여분의 염색액을 슬라이드에서 제거한다.
다음, 저배율(4×10)로부터 고배율(10×40)로 관찰한다.
제8단계 : 체세포의 핵상관찰(Giemsa Stain 이용)
Giemsa stain을 이용하여 체세포의 핵상관찰을 위해 다음 준비물을 사용하였다. 즉, 저장처리용액 (0.9% sodium citrate 용액 또는 0.56% KCL 용액); 고정액 (메탄올 : acetic acid = 3 : 1); 5% Giemsa; 슬라이드 글라스; 15ml 시험관 체세포의 관찰과정으로는, 디쉬내의 세포를 부유시킨 후(Single cell), 15ml 시험관에서 1500rpm으로 2분간 원심분리하여 세포 펠릿(pellet)을 얻는다.
다음, 펠릿(pellet)에 저장액을 첨가하고 부유하여 15분간 수화시킨 다음 총 부피의 1/3에 해당하는 고정액을 첨가한다.
그 후, 원심분리시킨(1500rpm, 2분) 다음, 고정액으로 세척하고(원심분리→ 상층액 제거→재부유, 3회), 마지막 원심 분리 후 적정농도가 되도록 고정액을 남겨 세포 부유 슬라이드위에 점적(최대한 확산하도록)시키고나서, 공기중에서 건조시킨다.
그리고 Giemsa 용액내에서 15분간 염색시킨 다음, 증류수를 이용하여 여분의 염색액을 슬라이드에서 제거한다.
그 후, 저배율(4×10)에서 고배율(10×40)로 관찰한다.
실시예 2 : 소의 핵이식
핵 이식준비 과정은 다음과 같다.
제1단계 : Micro 피펫의 준비
Micro피펫을 준비함에 있어서 사용기기는,Puller(Narishige PC-10), Microforge(Narishige MF-9), Glinder(Narishige EG-4) 등이다.
우선, 절개 및 탈핵용 피펫(cutting Enucleation 피펫)제조하기 위하여, 유리관(Drummond; Sigma, P-2174 or narishige)을 사용하여 추(Light 1개와 heavy 1개 또는 light 2개, heavy 1개)를 부착하고 Pulling step으로 Step 2에 고정한 다음, 온도를(heat 1- 101, heat 2- 101.5) 조정한다.
두번째로, 공여핵세포 이식용 피펫(Injection pipette, 내경 30∼40㎛)제조하기 위하여는 유리관(Drummond; Sigma,P-2174 or narishige)을 사용하여 추(Light 1개) 또는 추를 제거하고 Pulling step으로 Step 2에 고정한 다음 온도를(heat 1∼101, heat 2∼2 101.5) 조정한다.
다음에, 고정용 피펫(Holding pipette, 내경 70∼130㎛)제조(2가지)하기 위 하여는 다음과 같이 하였다.
i) 유리관(Drummond; Sigma, P-2174)를 사용하는 경우 :추(Light 1개 ∼light 2개, heavy 1개)를 부착하고 Pulling step(Step 1)에 고정한 다음 온도를 heat 2∼101.5으로조정한다.
ii) 유리관(Narishige G-1 or GD-1)을 사용하는 경우추(Light 1개, heavy 1개)를 부착하고 Pulling step(Step 1)에 고정한 다음, 온도는 heat 2∼101.5로 조정한다.
넷째로, 난자의 세정 및 운반용 피펫 제조하기 위하여알코올 램프를 이용하여 내경이 300㎛ 이상이 되도록 하고, 사용시 한번 이용한 것은 폐기한다.
다섯째로, 난구세포제거(Denuding용)용 피펫을 제조하기 위하여는 알코올램프를 이용하여 내경이 약 130㎛인 것을 여러개 만들어 준비한다.
제2단계 : 배양액의 조제
배양액(medium)은 다음과 같이 준비하였다.
IVM용 TCM-199으로 체외수정과 동일하게 제조하고, TCM-W으로, 사용 당일에는 작업용 미소적을 작성하되 9ml의 TCM-W에 1ml의 FBS(FCS)를 넣어 10%FBS TCM-W를 만든 후, 필요한 점적 수만큼 100㎕ 피펫을 이용하여 20∼30㎕ 점적을 적정한 크기(35mm, 60mm, 100mm등)의 디쉬에 만들고, 미네랄 오일로 도포한 다음 사용한다.
또, 하이알루로니데이즈(Sigma, H-3506) solution을 제조하기 위해서는, 5ml의 TCM-W에 하이알루로니데이즈(Hyaluronidase) 0.0500g을 첨가하여 원액(stock)을 만든다. 만들어진 원액은 111㎕씩 eppendorf 시험관에 분주하여 냉동실에 보관한다.
사용시에는 TCM-W를 1ml 분주하여 최종농도를 0.1%로 맞추어 35mm 디쉬에 분주하여 난구세
포제거(denuding)에 이용한다.
그리고, Cytochalasin B(sigma, C-6762) solution 제조를 위해서는, DMSO(Sigma, D-5879) 133㎕에 1mg의 사이토칼라신 B(cytochalasin B)를 녹이면 농도는 7.5mg/ml이 되는데, 이것을 eppendorf 시험관에 1㎕씩 분주해서 냉동실에 보관한다.
사용할때는 여기에 10% FBS(또는 FCS)첨가 TCMW를 1ml 첨가하여 절개 및 탈핵(cutting enucleation) 미소적 제작에 이용한다.
또한, PHA-P(Phytohematoglutinin: L-9132) solution 제조법은 5mg의 PHA를 10ml의 TCM-W와 섞은 다음 100㎕씩 eppendorf 시험관에 분주한 다음, 실험에 사용할 경우 eppendorf 시험관에 400㎕의 TCM-W를 첨가하여 작업용 미소적을 만들어 사용한다.
이때 최종 농도는 100㎍/ml이 되도록 한다.
그리고, 전기융합 및 활성화 배지(Electrofusion and Activation media) 제조를 위해서는 Mole 농도에 따른 만니톨(Sigma, M-1902)의 첨가량 (100ml기준)을 0.28M 만니톨: 5.1016g; 0.25M 만니톨: 4.555g; 0.26M 만니톨: 4.7372g; 0.1mM MgSO 4 , 0.1mM CaCl 2 포함하고, 기타 첨가물(만니톨 mole 농도에 관계없음): 0.1mM MgSO 4 또는 MgCl 2 , 0.1mM CaCl 2 (융합과 활성 동시군에서 첨가); Hepes 0.5mM; BSA 0.05%을 첨가하되 DW를 100ml까지 첨가한다.
pH를 7.2∼7.4로 맞추고, 5ml 시험관에 약 4ml씩 분주하여 냉동(장기간) 혹은 냉장실(단기)에 보관한다.
한편, DMAP(Sigma, D-2629)제조법은 10ml IVC용액에 0.0031g DMAP을 첨가하여 용해 및 여과후(0.22㎛ filter사용) eppendort 시험관에 분주시키고 냉동보관(1.9mM)한다.
또, Cycloheximide(Sigma, C-7698)는, Ethanol 100ml에 Cycloheximide 1g을 녹여 ×1000배 Stock을 작성 최종 농도10㎍/㎖가 되도록 IVC medium에 첨가하여 사용한다.
그리고, Ionomycin(Sigma, I-0634)는, DMSO(Sigma, D-5879) 1.34ml에 1mg의 Ionomycin를 녹여 1mM 원액을 제조한 다음, 최종농도 5㎛이 되도록 1% BSA TCM-W 또는 IVC medium내에 첨가하여 사용하고 원액을 냉동보관한다.
Ca 2+ Ionophore(A23187)는, DMSO(Sigma, D-5879) 1.66ml에 1mg의 Ca 2+ Ionophore(A23187)를 녹여 1mM원액 제조시 최종농도 5㎛가 되도록 1% BSA, TCM-W 또는 IVC medium내에 첨가하여 사용하고 원액을 냉동보관한다.
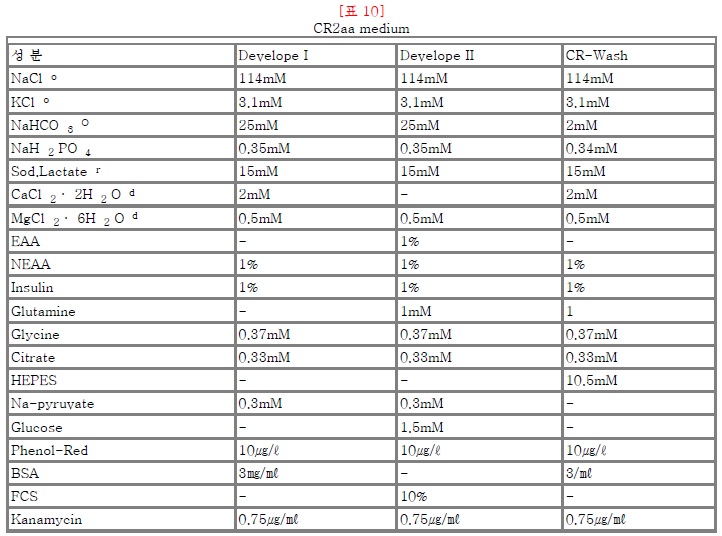
배양액(culture Medium)은 mTALP (No. 11), mSOF (No. 12), CR2aa (No. 13), 난관공배양(No. 14)중에서 선택하고, Hoechst 33342 (Sigma, B-2261)는 25mg의 Hoechst에 증류수를 5ml 섞어 희석(5mg/ml 의 stock)시킨 다음, Stock 1∼2㎕씩 eppendorf 시험관에 분주, 냉동보관한다. 그리고, 사용시에는 시험관에 10% FBS 첨가하고 TCMW를 1ml 첨가하여 희석시킨 후 사용(최종농도 5∼10㎍/ml)한다.
10∼15분간 정치한 다음 암실, 형광현미경으로 탈핵여부를 판단할 수 있다.
Nocodazole stock (Sigma, M-1404)은, DMSO(Sigma, D-5879) 2.5ml를 10mg의 nocodazole병에 넣어 희석시킨 다음 50∼100㎕씩 eppendorf 시험관에 분주(4mg/ml의 stock)하고 Stock 1㎕를 원하는 배양액 10ml에 첨가(0.4㎍/m)한다.
Acid Tyrode's(Sigma, T-1788) 제조법은 Embryo tested된 완제품으로 미소적(25㎕)을 만들어 오일도포 후 사용한다.
제3단계 : 미분간섭 현미경 작업용 디쉬 제작
미분간섭(Differential Interference Contrast, DIC)현미경 작업용 디쉬는 다음과 같이 제작하였다.
50mm 디쉬(Falcon 1006) 뚜껑에 직경 1∼3cm로 천공한 다음 Silicon (ShinEtsu KE45)을 디쉬 내부의 구멍주위에 도포한 후, 가로×세로 크기가 2∼4cm의 커버글라스를 실리콘위에 놓고 눌러서 부착시킨다.
끝으로, 알코올로 세정한다.
제4단계 : DIC 현미경용 미소적 제작
DIC 현미경용 미소적 제작법은 다음과 같이 제작하였다.
DIC용 작업용 디쉬의 커버글라스 위에 20㎕ 피펫을 이용하여 5∼10㎕ 미소적을 세로로 분주한 다음(미소적의 개수는 난자 20∼30개당 1개씩 계산) 1㎖ 피펫을 이용하여 4㎖정도의 미네랄 오일(sigma, M-3516)을 미소적 주위에 도포한다.
이때, 각 과정에 적합한 배지로 미소적을 형성시킨다.
제5단계 : 공여핵원용 체세포 준비
공여핵원용 체세포(Somatic cell line for donor)의 준비는 다음과 같이 수행하였다.
Tissue에서 세포주를 분리한다(다음 실험예 참조).
실험예 1 : 자궁에서의 세포주 분리
자궁관류액으로부터의 세포 채취 과장은 먼저, 자궁관류액을 원심분리 시험관에 옮긴 후, 인산염 완충 식염수(PBS) (GIBCO BRL, Life Technologies, USA)에 0.5% P/S항생제(penicillin 10000IU, streptomycin 10mg)를 첨가하여 1500rpm으로 2분간 원심분리한다.
다음, 원심분리 후, 상층액을 버리고 다시 상기의 PBS를 첨가한 후, 피펫을 사용하여 침전된 세포를 재부유시킨다.
재부유한 세포를 다시 1500rpm으로 2분간 원심분리하며, 이 과정을 3∼5회 반복한다.
최종 원심 세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨가 DMEM(Dulbecco's modified Eagles medium, GIBCO BRL, Life Technologies, USA) 에 부유하여 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2 ,포화습도 배양기내에서 배양한다. 세포가 배양접시 저면에 부착하여 분열 증식하여 단층(monolayer)을 형성하면 이를 1차 세포주(primary cell line)의 성립으로 간주한다.
자궁내막에서의 세포 채취 과정은먼저, 도축장에서 자궁을 채취하여 청결한 상태로 실험실로 운반한다.
다음, 자궁난관 접합부에서 10cm되는 부위를 멸균된 기구로 횡단한다.
횡단면을 집게로 보정한 후, 가위로 3cm 정도 횡단면에서 난관쪽으로 수직 절개한다.
절개 후, 수술용 날, 혹은 스크레이퍼(scraper) 등으로 자궁내막을 긁어낸다.
그리고, 채취된 내막의 일부를 1)의 PBS에서 2∼3회 세정 후 0.25% 트립신, 1 mM EDTA 용액(GIBCO BRL, Life Technologies, USA)으로 옮겨 39℃, 5% CO 2 의 포화습도 배양기내에서 30분 정치시킨다. 30분간 정치시킨 후, 와동(vortexing) 혹은 피펫을 사용하여 하여 자궁관류액으로부터의 세포 채취과정과 같은 조건하에 원심분리시킨다.
상층액을 버리고 침전된 조직 및 세포에 상기 자궁관류액으로부터의 세포 채취과정의 PBS를 첨가하여 재부유시킨 후 다시 원심분리한다.
이 과정을 3∼5회 반복한다.
최종 원심세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨 가 DMEM(Dulbecco's modified Eagles medium, GIBCO BRL, Life Technologies, USA)에 부유시킨 후, 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2의 포화습도 배양기내에서 배양한다.
세포가 배양접시 바닥에 부착하여 분열 증식하여 단층(monolayer)을 형성하면 이를 1차 세포주의 성립으로 간주한다.
실험예 2 : 난관에서의 세포 분리
도축장에서 낭종 등이 없는 정상적인 난소를 선별하여 난관채와 자궁난관 결합부를 포함한 난관을 자궁간막에서 절제하여 청결한 상태로 실험실로 운반한 다음 상기 실험예 1의 PBS 세정 후에, 난관 주변조직을 가위 등으로 절제한다
.
다음 멸균된 슬라이드 글라스를 준비하고, 난관채에 인접한 부위를 0.5cm절제 후, 멸균된 슬라이드 글라스위에 고정시킨다.
여분의 슬라이드 글라스의 날을 세워 난관의 중간부위에서 난관채쪽으로 밀어낸다.
밀려나온 난관상피 조직과 세포를 슬라이드 글라스에서 원심분리용 시험관으로 옮기고, 상기 실험예 1의 PBS를 첨가하여 부유한 후 1의 조건에서 원심분리한다.
상기 실험예 1과 같은 방법으로 원심 세정한다.
최종 원심세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨 가 DMEM(Dulbecco's modified Eagles medium, GIBCO BRL, Life Technologies, USA)에 부유시킨 후, 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2의 포화습도 배양기내에서 배양한다.
세포가 배양접시 바닥 저면에 부착하여 분열 증식하여 단층을 형성하면 이를 1차 세포주의 성립으로 간주한다.
실험예 3 : 난구세포(Cumulus cell)의 분리
먼저, 난구난자세포 복합체(Cumulus-oocyte complex)를 하이알루로니데이즈 (Hyaluronidase)로 처리하여 난자를 제외한 난구세포층을 분리(체세포 핵이식 참조)한 다음 난구세포층에 상기 실험예 1과 같은 트립신(trypsin)-EDTA용액을 첨가하여 39℃, 5% CO 2 의 포화습도 배양기내에 30분 내지 1시간 정치시킨다.
트립신-EDTA처리 후 와동(vortexing) 및 피펫팅 후 상기 실험예 1과 같은 조건에서 원심세정한다.
최종 원심세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨가 DMEM(Dulbecco's modified Eagles medium, GIBCO BRL, Life Technologies, USA)에 부유시킨 후, 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2의 포화습도 배양기내에서 배양한다.
세포가 배양접시 바닥 저면에 부착하여 분열 증식하여 단층을 형성하면 이를 1차 세포주의 성립으로 간주한다.
실험예 4 : 귀(ear)에서의 세포 분리
성체 또는 어린 동물의 귀 선단에서 떼어낼 부위 주변의 체모를 깎고, 소독용 알코올과 베타딘으로 소독한다.
멸균된 기구로 1∼2cm 2 넓이의 피부 조직을 절제한 다음, 50 ml 시험관에 상기 실험예 1의 PBS용액을 준비하여 절제된 조직을 넣어 4℃로 냉장 보관하여 실험실로 운반한다.
다시 상기 실험예 1의 PBS 용액으로 세정한 다음, 멸균된 집게로 고정한 후, 체모를 포함한 피부와 연골조직을 멸균된 가위, 날(blade)로 분리하여, 연골조직과 면한 피부 내측의 조직을 채취한다.
조직편을 다시 상기 실험예 1의 PBS 용액으로 세정 후 칼날(blade)로 세절한 다음, 상기 실험예 1의 0.25% 트립신,1mM EDTA 용액과 0.8% 콜라게나제형 (Collagenase type III) II(GIBCO BRL, Life Technologies, USA) 용액의 혼합액을 첨가하여 39℃, 5% CO 2 의 포화습도 배양기내에서 배양한다.
정치 후 원심 분리용 시험관에 옮겨 와동(vortexing)과 피펫작업 (pipetting) 후, 상기 실험예 1)의 PBS 용액을 첨가하여 같은 조건으로 원심세정한다.
최종 원심세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨가 DMEM(Dulbecco's Modified Eagles medium, GIBCO BRL, Life Technologies, USA)에 부유시킨 후, 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2의 포화습도 배양기내 에서 배양한다.
세포가 배양접시 바닥면에 부착하여 분열 증식하여 단층이 형성되면 이를 1차 세포주의 성립으로 간주한다.
실험예 5 : 근육에서의 세포 분리
생검(biopsy)에 의해 혹은 신선 사체 등에서 채취한 골격 및 동체의 근육 조직을 1 cm 3 크기로 절제하여 실험예 4와 같은 방법으로 실험실로 운반한 다음, 조직편을 다시 상기 실험예 1의 PBS 용액으로 세정한 후, 멸균 가위와 칼날(blade)로 세절한다.
상기 실험예 1의 0.25% 트립신, 1mM EDTA 용액과 0.8% 콜라게나제형 II(GIBCO BRL, Life Technologies, USA)용액의 혼합액을 첨가하여 39℃, 5% CO 2 의 포화습도 배양기내에서 배양한다.
정치 후 원심 분리용 시험관에 옮겨 와동(vortexing)과 피펫작업 (pipetting) 후, 상기 실험예 1의 PBS 용액을 첨가하여 같은 조건으로 원심세정한다.
최종 원심세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨가 DMEM(Dulbecco's Modified Eagles medium, GIBCO BRL, Life Technologies, USA)에 부유시킨 후, 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2의 포화습도 배양기내에서 배양한다.
세포가 배양접시 바닥면에 부착하여 분열 증식하여 단층이 형성되면 이를 1차 세포주의 성립으로 간주한다.
실험예 6 : 태아섬유아세포(fetal fibroblast)의 분리
먼저, 임신자궁에서 무균적으로 임신 30 내지 60일령의 태아를 채취한다.
양막을 포함한 태아를 상기 실험예 4와 같은 방법으로 실험실로 운반한다. 상기 실험예 1의 PBS 용액으로 수차례 세정한다.
두부와 내장 부위를 제거하고 동체와 사지부위에서 연골 조직을 포함하지 않는 부위의 조직을 채취, 멸균 가위와 칼날(blade)로 세절한다.
상기 실험 예 1의 0.25% 트립신, 1mM EDTA 용액과 0.8% 콜라게나제형 II(GIBCO BRL, Life Technologies, USA)용액의 혼합액을 첨가하여 39℃, 5% CO 2 의 포화습도 배양기내에서 배양한다. 정치후 원심 분리용 시험관에 옮겨와동 (vortexing)과 피펫작업 (pipetting) 후, 상기 실험예 1의 PBS 용액을 첨가하여 같은 조건으로 원심세정한다.
최종 원심세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨가 DMEM(Dulbecco's Modified Eagles medium, GIBCO BRL, Life Technologies, USA)에 부유시킨 후, 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2의 포화습도 배양기내에서 배양한다.
세포가 배양접시 바닥면에 부착하여 분열 증식하여 단층이 형성되면 이를 1 차 세포주의 성립으로 간주한다.
이하, 분리된 세포주의 유지를 위하여 다음과 같이 조치하였다.
실험예 7 : 계대 배양
단층이 형성된 1차 세포주는 계대배양을 다음과 같이 실시한다.
페트리디쉬의 배양액을 버리고, 상기 실험예 1의 0.25% 트립신, 1mM EDTA 용액을 첨가하여 1 내지 2분 정치 후, 배양용 배지로 교체한다.
피펫팅하여 세포를 배양 배지 중에 재부유시킨 다음 적당량을 새로운 페트리디쉬에 분주한 후, 배지를 첨가하여 39℃, 5% CO 2 의 포화습도 배양기내에서 배양한다.
실험예 8 : 혈청기아(serum starvation) 배양
상기 실험예 1의 10% FCS 1% 비필수 아미노산 용액, P/S 첨가 DMEM (Dulbecco's Modified Eagles Medium, GIBCO BRL, Life Technologies, USA)에서 배양, 증식중인 세포주을 0.5 % FCS 첨가 배양액으로 교체한다.
3 내지 10일간 배양 후 공여핵원으로 제공한다.
실험예 9 : 세포주의 동결보존
유착상태의 세포를 상기 실험예와 같은 방법으로 재부유하여 세포 수가 1 X 10 5 /ml로 되도록 조절한 다음, 세포를 포함한 전체 배양액 중에 DMSO(Dimethyl sulfoxide, Sigma, USA)가 10% 농도가 되도록 첨가하여 희석한 후 Cryovial (Corning, USA)에 봉입한다.
Cryovial을 -70℃ 냉동고에 넣어 18∼24시간 보관한 후 액체 질소 탱크에 넣어 보관한다.
실험예 10 : 동결보존 세포주의 융해액체질소 탱크에서 Cryovial 을 꺼내어 37℃ 온수에 넣어 융해시키고, 융해된 세포를 원심 분리용 시험관에 넣고 상기 실험예 1의 PBS를 첨가하여 1,500rpm, 2분간 원심 세정한다. 상층액을 버리고 세포 침전물을 조심스럽게 하여 원심 세정을 2 회 반복한다.
최종 원심세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨가 DMEM(Dulbecco's Modified Eagles medium, GIBCO BRL, Life Technologies, USA)에 부유시킨 후, 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2의 포화습도 배양기내에서 배양한다.
실시예 3 : 체세포 핵이식
제1단계 : 체외성숙(In vitro maturation)
상기 실시예 1의 체외성숙에서와 같은 방법으로 수핵란을 채취한다.
제2단계 : 난구세포 제거(Denuding)
준비물(Preparation)은 다음과 같다.
세정용 피펫, 제거용 피펫, 하이알루로니데이즈, TCM-W 작업용 미소적 작업과정(Procedure)은 다음과 같다.
IVM 16∼22시간의 난자를 세정용 마우스 피펫을 이용하여 TCM-W에 1회 세정한다.
하이알루로니데이즈(No. B-③)용액내에 난자를 분주한다.
제거용 피펫으로 난구세포를 벗긴다. TCM-W에 3회정도 세정하고 정치시킨다. 상기 작업은 신속하게 수행하는 것이 바람직하다.
제3단계 : 절개 및 탈핵
준비물(Preparation)은 다음과 같다.
작업용 디쉬(No. C), 작업용 미소적 - 사이토칼라신 B(Cytochalasin B), 피펫 : 고정용 피펫, 절개 및 탈핵용(cuttingEnucleation) 피펫 절개 및 탈핵과정(Procedure)은 다음과 같다.
작업용 디쉬를 조작판(manipulator plate)위에 위치하도록 놓는 다음, 피펫을 장착한다.
조작장치(Manipulator)의 왼쪽 암(arm)에 고정용 피펫을 장착한 후 조작장치의 오른쪽 암(arm)에 절개용 피펫을 장착한다.
피펫의 방향 및 위치를 예컨대 다음과 같이 조정한다.
즉, 고정용 피펫은 9시 방향에 위치시키고 절개용 피펫은 3시 방향에 위치시킨다. 피펫 조정기를 중립에 놓아 피펫이 상하 좌우로 자유롭게 움직일 수 있도록 조정한다. 두 피펫을 작업용 점적내로 상하 유동시키면서 피펫이 디쉬의 테두리에 닿지 않도록 각도를 조정하고 피펫 끝이 미소적의 중앙에 위치시킨다.
세정용 마우스 피펫(내경 200㎛이상)을 이용하여 TCM-W에 있던 난자(oocyte) 10∼30개 정도를 1개 군으로 하여 절개 및 탈핵 후 점적으로 이동시킨다.
다음 방식으로 절개를 실시한다:
즉, 조동나사와 미동나사를 이용하여 난자(oocyte)에 먼저 초점을 맞춘 뒤, 2개의 피펫을 상하로 움직여 초점을 조정한다.
2개의 피펫을 움직여서 파지용 피펫의 12시 방향에 제 1극체(The first polar body)가 위치하도록 한다.
파지용 피펫을 난자(oocyte)의 9시 방향에 밀착시킨 뒤 유압을 걸어 난자(oocyte)를 고정시킨다. 절개 피펫을 1시 방향에서 찔러서 투명대를 통과시킨 뒤 세포질이 다치지 않도록 주의하며 11시 방향으로 관통시킨다.
파지용 피펫에 양압을 걸어 난자를 분리하고 파지용 피펫을 절개 피펫이 통과한 제 1극체 상단부의 투명대에 접촉시키고 두 피펫을 마찰하여 투명대를 절개한다.
탈핵(Enucleation; Squeezing method)은 다음과 같이 수행한다.
절개한 난자를 회전시켜 절개창을 수직으로 위치시킨 후 고정용 피펫을 난자의 밑 부위에 위치시켜 난자가 아래쪽으로 움직이지 못하도록 받친 뒤 절개 피펫을 난자의 위에서 눌러 제 1극체를 포함하여 세포질을 10∼30% 탈핵을 실시 한다.
또는 난자의 절개창을 3시방향에서 수직으로 회전시킨 후 고정용 피펫과 절개 피펫을 아래 위에서 눌러서 제 1극체를포함하여 세포질의 10∼30% 탈핵을 실시한다.
탈핵시킨 1군의 난자는 TCM-W로 3회 세정하고 핵이식 전까지 IVM용 TCM 199(B-①)에 정치시킨다.
탈핵 후의 난자는 매우 취약하기 때문에 마우스 피펫은 최소한 내경이 300㎛이상되는 것을 사용하여 작업후 난자의 세포질이 절개창을 통해 빠져 나오지 않도록 주의한다.
제4단계 : 공여핵원용 단일 세포의 분리
페트리 디쉬내의 배양액을 버리고 상기 실험예 1의 0.25% 트립신, 1mM EDTA 용액을 첨가하여 1∼2분 정치시킨다.
정치 후, 1% FCS 첨가 PBS(-)(GIBCO BRL, Life Technologies, USA)에 피펫팅에 의해 재부유시킨다.
세포의 수가 적당량이 되도록 조절하여 (2,000/0.1ml) Eppendorf 시험관에 분주시킨다.
제5단계 : 공여세포 이식(Injection)
준비물(Preparation)은 다음과 같다.
작업용 디쉬, 작업용 점적 만들기-PAH, 피펫 : 고정용 피펫, 주사용 피펫, 공여 세포 미소적(Donor 세포 준비 참조)작업과정(Procedure)은 다음과 같다.
작업용 디쉬를 조작판(manipulator plate)에 위치시킨다.
절개 피펫을 주사용 피펫으로 교체시킨다.
IVM 배지내에 있는 난자(oocyte)를 세정용 마우스 피펫을 이용하여 TCM-W 점적에서 1∼3회 세정후 주입용 미소적으로 이동시킨다.
이식용(Injection) 피펫을 공여세포 미소적에 넣고 20∼40㎛크기의 1개 이상의 공여세포를 흡입한 뒤 피펫을 주입용 미소적으로 이동시킨다.
절개창을 양 피펫과 1시방향에 수직으로 놓고 고정용 피펫으로 고정한다.
이식용(Injection) 피펫을 절개창으로 주입하고, 유압을 걸어 1개의 공여세포을 주입시킨다.
작성된 핵이식란 군을 마우스 피펫으로 세정용 미소적으로 옮겨 1∼3회 세정하고 정치시킨다.
실시예 4 : 세포융합(Cell Fusion)
세포융합에 필요한 준비물(Preparation)은 기기-BTX ECM 2001의 다음과 같다.
만니톨(Manitol)을 4-웰 디쉬의 3개의 웰(1번, 2번, 3번 웰, 순서는 상관없음)에 0.4ml∼1ml 분주하고, 나머지 1개의웰 (4번)에는 10% TCM-W가 첨가된 만니톨을 제조한다.
만일 융합과 활성화를 동시에 실시할 경우는 Ca 2+ 첨가 융합배지를 사용하 며, 융합후 활성화군에서는 Ca 2+ 를 배제한 융합배지를 사용한다.
다음, 2개 전극(3.2mm chamber No. 453)에 만니톨을 0.2∼1ml 넣고 BTX-세포 조작기와 연결시킨다.
이때, 세포융합 조건은 다음과 같다.
DC 조건 : 전압(Voltage)은 0,75∼2.25kv/cm이며, 횟수(Times)는 1∼2회, 시간(Seconds)은 회수×시간 총 60㎲ 이내로 조정한다.
[태아섬유아세포(Fetal fibroblast) - DC 1.75∼1.85KV/cm, 20㎲, 1회]
(Ear 세포 - DC 1.75∼1.8KV/cm, 15㎲, 1sec 간격, 2회)
[자궁(Uterine) 세포 - 1.85∼2.25KV/cm, 15㎲, 1sec 간격, 2회]
작업과정(Procedure)은 다음과 같다.
작업 1시간전 BTX를 가동하여 예열한 다음 세정용 마우스 피펫에 10∼20㎕의 만니톨을 빨아들인 후 핵이식란이 있는 TCM-W 미소적에 첨가한다.
1분 이상 정치시킨 후 세정용 마우스 피펫을 이용하여 4번 웰(10% TCM-W 만니톨)에 핵이식란을 주입한다.
1분 이상 정치시킨 후 세정용 마우스 피펫을 이용하여 1번 웰에 넣고, 같은 방법으로 2번 웰에 핵이식란을 이동시킨다.
마우스 피펫을 이용하여 핵이식란을 하나씩 2개 전극 사이에 놓고 공여세포가 (+)전극을 향하도록 정렬시킨다.
통전을 실시하고 융합한 핵이식란은 3번 웰에 모아서 1개 군의 융합이 끝나 면 TCM-W로 옮겨 3회 세정한다.
Cytochalasin B 미소적내에서 30∼60분간 처리후, 융합·활성을 동시에 실시하게 될 군은 후활성화(postactivation)과정으로, 융합 후에 활성화시킬 군은 상기의 화학물질을 이용하여 활성화시킨다.
활성화(Activation)는 다음과 같이 실시한다.
융합과 동시에 일렉트로퓨젼(elecrofusion) 방법으로 활성화를 유도하는 경우에는 Ca 2+ 첨가 융합배지내에서 일렉트로퓨젼(electrofusion)을 실시한다.
Ionomycin(B-⑨)처리법으로 활성화를 유도하는 경우에는, B-⑨, 50㎕ 점적내에서 4분간 정치(빛에 민감하므로 되도록 빛의 유입을 차단)하고, 35mm 디쉬내 10% FBS 혹은 30mg/ml BSA 첨가하고 세정용 매질하에서 이노마이신 (Ionomycin)을 제거한 뒤 5분간 정치시킨다.
실시예 5 : 융합 후 활성화(Postactivation)
Cycloheximide 25㎕ 미소적 내에 5∼10개 정도의 작성란을 침지하여 6시간 배양한다.
DMAP 25㎕ 미소적 내에 5∼10개 정도의 작성란을 침지하여 4시간 배양시킨다.
배양은 다음과 같이 한다.
상기 실시예 2 제2단계의 배양액(Culture medium) 중 택일하여 5% CO 2 배양 기 또는 5% CO 2 , 7% O 2 , 88%N 2 에서 5∼8일간 배양한다. 활성화(Activation) 후 24시간 전후로 검란,발육단계별로 구분하여 미소적내에 배치 한다.
최종 검란은 수정 7일에 실시하여 후기배 발육율을 계산하고 I-4, 5의 방법으로 핵이식란의 동결 및 이식을 실시한다
.
이상과 같이, 소의 체외수정 및 체외성숙에 의해 얻어진 수정란 또는 소의 체세포를 채취하여 세정 및 분주처리조건하에 보관하고, 소의 다양한 조직으로부터 세포주를 분리보존하고, 최초 세포의 세포막의 일부를 절개하고 세포질의10 내지 30%의 탈핵을 실시한 다음, 상기 공여세포를 상기 핵이 탈핵된 난자에 이식하여 전기융합시키고 활성화 및
후활성화 단계 후, 배양 및 성숙시킴으로써, 체세포의 복제에 의해 소를 생산할 수 있게 된다.
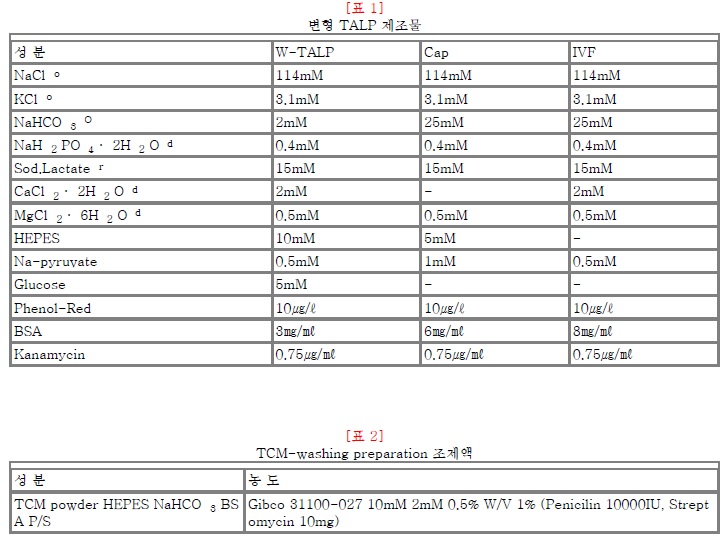
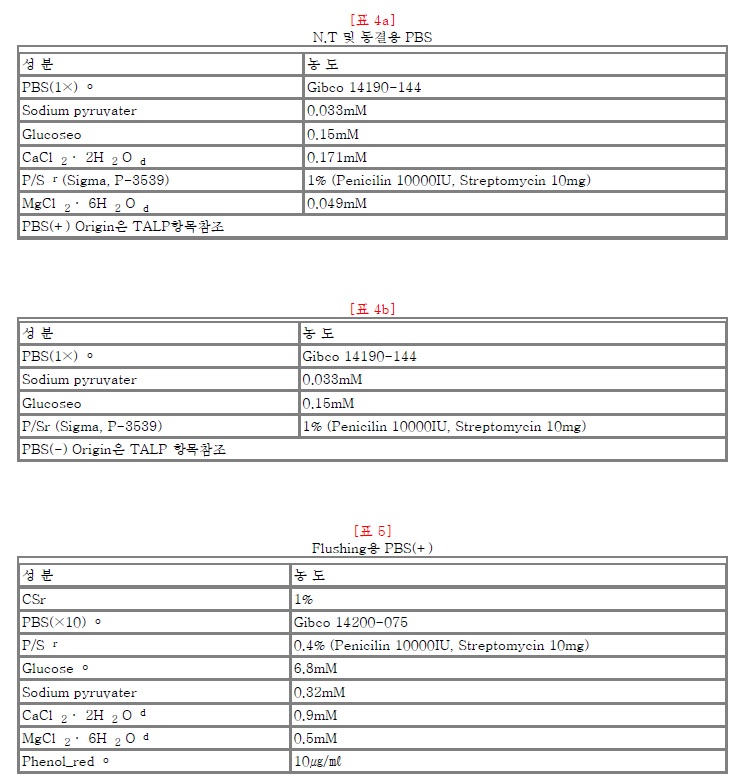
이하에서는 전술한 본 발명의 각각의 공정에 사용되는 다양한 배지들의 종류와 그 조성을 제시한다:
표1)

소 항생제(bovine antibiotics)는 다음과 같다.
P/S bottle(Sigma, Penicillin 10000IU, Streptomycin 10mg)에 20ml DW를 첨가
FSH(Antrin, 일본 Denka)/ Estradiol(Sigma, E-8875)은 다음과 같다.
1. TCM-C 5㎖를 1 vial에 넣고(10㎎/5㎖)
2. 1㎖를 취한다. -(2㎎을 취한 셈)
3. 딴 1㎖를 TCM-C 9㎖와 합친다.
(2㎖/10㎖) ⇒ 200㎕씩 eppendorf 시험관에 분주하여 냉동보관 Estradiol : 1㎍/ml의 농도로 맞추려면 10ml의 에탄올에 5mg의 estradiol을 희석하고 1㎕를 0.5ml의 4-웰 디쉬에
첨가하면 된다.
이 때 에탄올이 증발하도록 빈 디쉬의 바닥에 estradiol을 먼저 분주하고 증발한 다음 FSH, 배양액등을 분주한다.
헤파린(200㎕/㎖)은 Cap-TALP 10ml에 0.002g 헤파린(sigma, H-3149)를 첨가하여 eppendorf 시험관에 300㎕씩 분주한후 냉동보관한다.
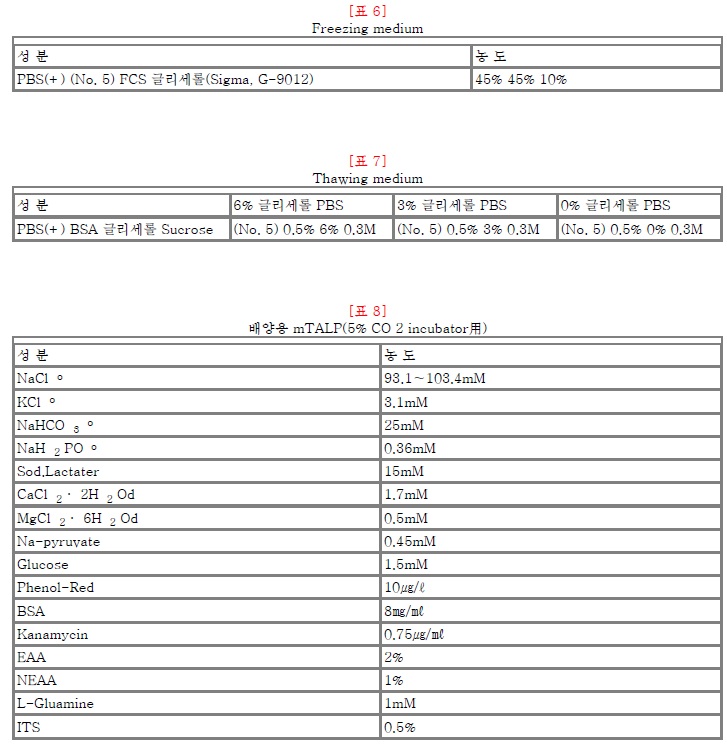
표6)
공배양은 다음과 같이 수행한다.
도축장에서 낭종 등이 없는 정상적인 난소를 선별하여 난관채와 자궁난관 결합부를 포함한 난관을 자궁간막에서 절제하여 청결한 상태로 실험실로 운반하여 DPBS로 세정 후, 난관 주변 조직을 가위 등으로 trimming하고 멸균된 슬라이드 글라스를 준비하여 난관채에 인접한 부위를 0.5cm절제 후, 멸균된 슬라이드 글라스위에 고정한 다음 여분의 슬라이드 글라스의 날을 세워 난관의 중간부위에서 난관채 쪽으로 밀어낸다.
밀려 나온 난관상피 조직과 세포를 슬라이드 글라스에서 원심분리용 시험관 로 옮기고, DPBS를 첨가하여 부유한 후 원심(1500rpm, 2분)분리한다.
DPBS로 2회 원심세정한 다음, 마지막 세정 후 펠릿을 5000∼10000개/ml의 세포농도가 되도록 TCM 199(No. 3)를 첨가하여 부유한다. 4웰 위의 부유액 500㎕을 분주, 배양하여 confluent monolayer(2×104)를 얻는다.
각 웰당 수정란 25∼50개를 첨가하여 공배양하게 되며 medium change는 2일 간격으로 실시한다.
실시예 6 : 자궁세포를 사용한 젖소의 복제('영롱이'의 경우) Holstein의 자궁관류액을 채취하여 원심분리 시험관에 옮긴 후, 인산염 완충 식염수(PBS) (GIBCO BRL, Life Technologies, USA)에 0.5% P/S항생제(페니실린 10000IU, 스트렙토마이신 10mg)를 첨가하여 1500rpm으로 2분간 원심 분리하였다.
원심분리 후, 상층액을 버리고 다시 상기의 PBS를 첨가한 후, 피펫을 사용하여 침전된 세포를 재부유시 켰다.
재부유한 세포를 다시 1500rpm으로 2분간 원심분리하며, 이 과정을 3∼5회 반복하였다.
최종 원심 세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨가 DMEM(Dulbecco's modified Eagles medium, GIBCO BRL, Life Technologies, USA)에 부유하여 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2 , 포화습도 배양기내에서 배양하였다. 세포가 배양접시 저면에 부착하여 분열 증식하여 융합(confluent)상태가 되었을 때,이를 1차 세포주(primary cell line)의 성립으로 간주하였다.
융합(confluent)상태를 확인한 후, 페트리디쉬의 배양액을 버리고, 0.25% 트립신, 1mM EDTA 용액을 첨가하여 2분정치 후, 배양용 배지로 교체하였다.
이를 피펫팅하여 세포를 배양 배지 중에 재부유시켰다. 적당량을 새로운 페트리디쉬에 분주한 후, 배지를 첨가하여 39℃, 5% CO 2 의 포화습도 배양기내에서 배양하였다.
이와 같이 계대배양을 실시하고, 후속의 배양 및 성숙단계를 지속적으로 수행하여 복제 소의 성장과 생육을 관찰 및 기록하고 '영롱이'라는 호칭을 부여하였다(도 1).
도1)

본 실시예의 방법으로 생산한 배반포 이상의 수정란은 SNU2(Bovine NT Embryo)로 명명하고 2000년 3월 10일 생명공학연구소 미생물기탁센터에 KCTC 0753BP로 기탁하였다.
실시예 7 : 소의 귀로부터 채취한 체세포를 사용한 한우의 복제('진이'의 경우)
어린 한우(도 3)의 귀 선단에서 떼어낼 부위 주변의 체모를 깎고, 소독용 알코올과 베타딘으로 소독하였다. 멸균된 기구로 1∼2 cm 2 넓이의 피부 조직을 절제하였다. 50 ml 시험관에 PBS용액을 준비하여 절제된 조직을 넣어 4℃로 냉장 보관하여 실험실로 운반하였다. 다시 PBS 용액으로 세정하였다.
멸균된 집게로 조직을 고정한 후, 체모를 포함한 피부와 연골조직을 멸균된 가위와 칼날로 분리하여, 연골조직과 면한피부 내측의 조직을 채취하였다.
조직편을 다시 PBS 용액으로 세정 후, 칼날(blade)로 세절하였다.
0.25% 트립신(trypsin), 1mM EDTA 용액과 0.8% 콜라게나제형 II(GIBCO BRL, Life Technologies, USA) 용액의 혼합액을 첨가하여 39℃, 5% CO 2 의 포화습도 배양기내에서 배양하였다. 정치 후 원심 분리용 시험관에 옮겨 와동 시키고 피펫작업 후, PBS 용액을 첨가하여 원심세정하였다.
최종 원심세정 후, 침전 세포를 10% FCS, 1% 비필수 아미노산 용액, P/S 첨가 DMEM(Dulbecco's modified Eagles medium, GIBCO BRL, Life Technologies, USA)에 부유시킨 후, 세포 배양용 페트리디쉬로 옮겨 39℃, 5% CO 2 의 포화습도 배양기내에서 배양하였다.
세포가 배양접시 저면에 부착하여 분열 증식하여 포화상태가 되었을 때 이를 1차 세포주의 성립으로 간주하였다.
이후에 상기 실시예 6과 동일한 과정을 거쳐 복제 소의 성장과 생육을 관찰 및 기록하고 '진이'라는 호칭을 부여하였다(도 2 및 도 3).
도2)

도3)

본 실시예의 방법으로 생산한 배반포 이상의 수정란은 Bos taurus coreanae embryos/TcEar로 명명하고 1999년 12월 31일 생명공학연구소 미생물기탁센터에 KCTC 0719BP로 기탁하였다.
발명자: 황우석, 이병천, 김성기, 신태영, 노상호, 박종임, 조종기, 김기연, 신수경, 송길영
대리인: 이덕록
|
권 리 란 |
|
표시번호 |
사 항 |
|
1번 |
|
출원 연월일 : |
2000년 06월 30일 |
출 원 번 호 : |
10-2000-0036742 |
|
우선권주장일자 : |
1999년 06월 30일 |
우선권 주장수 : |
1 |
|
우선권 주장국 : |
대한민국 |
|
공고 연월일 : |
2004년 01월 13일 |
공 고 번 호 : |
|
|
특허결정(심결)연월일 : |
2003년 12월 10일 |
청구범위의 항수 : |
4 |
|
유 별 : |
C12N 5/16 |
|
발명의 명칭 : |
체세포 복제동물 및 그 생산방법 |
|
존속기간(예정)만료일 : |
2020년 06월 30일 |
|
2003년 12월 22일 등록 | |
|
특 허 권 자 란 |
|
순위번호 |
사 항 |
|
1번 |
|
(등록권리자) |
|
황우석 |
|
|
서울시 강남구 논현동 ** 논현아파트 ***-*** |
|
|
2003년 12월 22일 등록 | |
|
2번 |
|
(권리의 전부이전등록) |
|
등록 의무자 : |
황우석 |
|
|
서울특별시 강남구 논현동 ** 논현아파트 ***-*** |
|
등록 권리자 : |
재단법인 서울대학교산학협력재단 |
|
|
서울특별시 관악구 봉천동 산*-*번지 |
|
등 록 원 인 : |
전담조직으로이전 |
|
등록의 목적 : |
권리의 전부이전 |
|
2004년 08월 07일 등록 | |
'세포 복제동물 및 그 생산방법' 특허 자세히 보기
 동향
동향
 화학 / 바이오 / 야금
화학 / 바이오 / 야금